

Population analysis of Legionella pneumophila reveals a basis for resistance to complement-mediated killing

Click to expand
December 9, 2021
Bryan A. Wee, Joana Alves, Diane S. J. Lindsay, Ann-Brit Klatt, Fiona A. Sargison, Ross L. Cameron, Amy Pickering, Jamie Gorzynski, Jukka Corander, Pekka Marttinen, Bastian Opitz, Andrew J. Smith & J. Ross Fitzgerald
Nat Commun. 2021 Dec 9;12(1):7165.
PMID: 34887398 | DOI: 10.1038/s41467-021-27478-z
Abstract
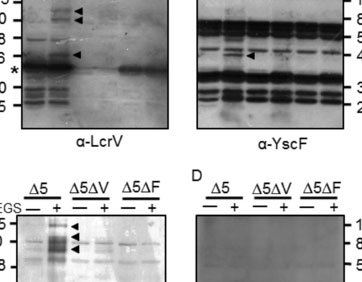
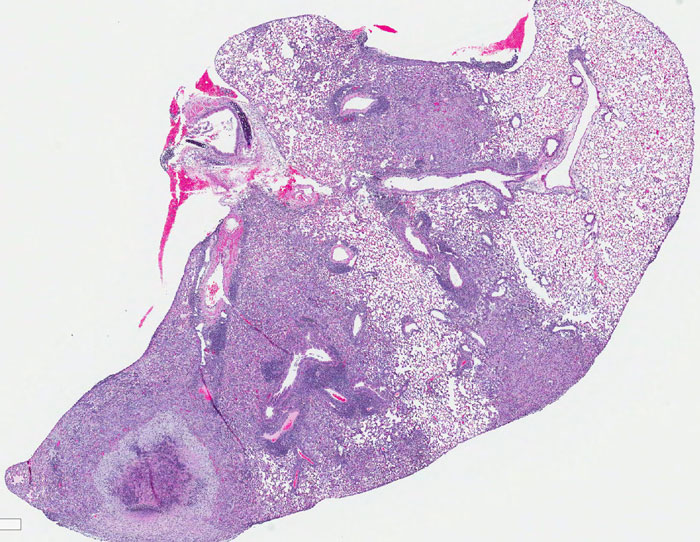
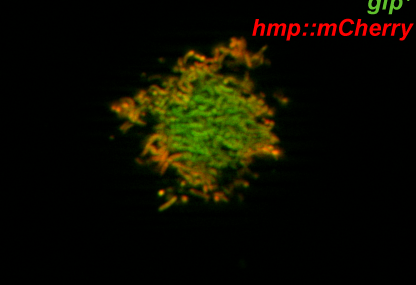
Legionella pneumophila is the most common cause of the severe respiratory infection known as Legionnaires’ disease. However, the microorganism is typically a symbiont of free-living amoeba, and our understanding of the bacterial factors that determine human pathogenicity is limited. Here we carried out a population genomic study of 902 L. pneumophila isolates from human clinical and environmental samples to examine their genetic diversity, global distribution and the basis for human pathogenicity. We find that the capacity for human disease is representative of the breadth of species diversity although some clones are more commonly associated with clinical infections. We identified a single gene (lag-1) to be most strongly associated with clinical isolates. lag-1, which encodes an O-acetyltransferase for lipopolysaccharide modification, has been distributed horizontally across all major phylogenetic clades of L. pneumophila by frequent recent recombination events. The gene confers resistance to complement-mediated killing in human serum by inhibiting deposition of classical pathway molecules on the bacterial surface. Furthermore, acquisition of lag-1 inhibits complement-dependent phagocytosis by human neutrophils, and promoted survival in a mouse model of pulmonary legionellosis. Thus, our results reveal L. pneumophila genetic traits linked to disease and provide a molecular basis for resistance to complement-mediated killing.